Mastering 2026 FDA MedTech Regulations: A Comprehensive Guide for US Professionals

Mastering the Latest FDA MedTech Regulations: A 2026 Educational Guide for US Professionals (RECENT UPDATES)
The medical technology (MedTech) landscape is in a constant state of evolution, driven by rapid innovation, emerging health challenges, and, critically, the ever-changing regulatory environment. For professionals operating within the United States, keeping pace with the U.S. Food and Drug Administration (FDA) regulations is not merely a formality but a cornerstone of successful product development, market entry, and patient safety. As we look towards 2026, several pivotal updates and reinforced guidelines are set to redefine how medical devices are designed, tested, manufactured, and monitored. This comprehensive guide aims to equip US MedTech professionals with the essential knowledge needed to navigate the FDA MedTech Regulations 2026, ensuring compliance and fostering innovation.
The FDA’s commitment to public health necessitates a dynamic regulatory framework. This framework balances the need for timely access to safe and effective medical devices with the imperative to adapt to scientific advancements and address new risks. Understanding these nuances is paramount for manufacturers, regulatory affairs specialists, quality assurance teams, and even investors in the MedTech sector. This article will delve into the significant changes expected, highlight areas of increased scrutiny, and provide actionable strategies to ensure your organization is not just compliant, but also strategically positioned for future success.
Our focus will primarily be on the FDA MedTech Regulations 2026, translating complex legal and scientific jargon into clear, actionable insights. We will explore key areas such as pre-market submissions, quality system requirements, post-market surveillance, and the escalating importance of cybersecurity in medical devices. Furthermore, we will touch upon the FDA’s increasing emphasis on real-world evidence and digital health technologies, which are rapidly reshaping the industry.
The Evolving Landscape of FDA MedTech Regulations 2026
The regulatory environment for medical devices is not static; it responds to technological breakthroughs, public health crises, and lessons learned from past experiences. The FDA MedTech Regulations 2026 are shaped by several overarching themes: enhancing patient safety, streamlining regulatory processes for innovative technologies, and adapting to the digital transformation of healthcare. These themes manifest in specific policy changes and enforcement priorities that MedTech companies must meticulously address.
Key Drivers of Regulatory Change
- Technological Advancements: The rapid pace of innovation in areas like artificial intelligence (AI), machine learning (ML), software as a medical device (SaMD), and personalized medicine continually challenges existing regulatory paradigms. The FDA is working to create pathways that accommodate these novel technologies without compromising safety and efficacy.
- Global Harmonization Efforts: While the FDA operates within the US, it actively participates in international harmonization initiatives (e.g., IMDRF, ICH). These efforts aim to align regulatory requirements globally, which can impact US regulations and facilitate international market access for US-based companies.
- Post-Market Performance and Real-World Evidence (RWE): There’s a growing emphasis on understanding device performance once it’s on the market. RWE, derived from electronic health records, claims data, and patient registries, is becoming increasingly critical for regulatory decision-making throughout a device’s lifecycle.
- Cybersecurity Threats: With the increasing connectivity of medical devices, cybersecurity has become a paramount concern. The FDA continues to strengthen its requirements for cybersecurity risk management, both pre-market and post-market.
- Supply Chain Resiliency: Lessons from recent global events have highlighted the fragility of global supply chains. The FDA is increasing its focus on supply chain transparency, quality, and resilience to prevent shortages and ensure continued access to essential medical devices.
Understanding these drivers provides context for the specific regulatory updates. For any MedTech professional, anticipating these shifts and proactively integrating them into their strategies is a hallmark of regulatory intelligence.
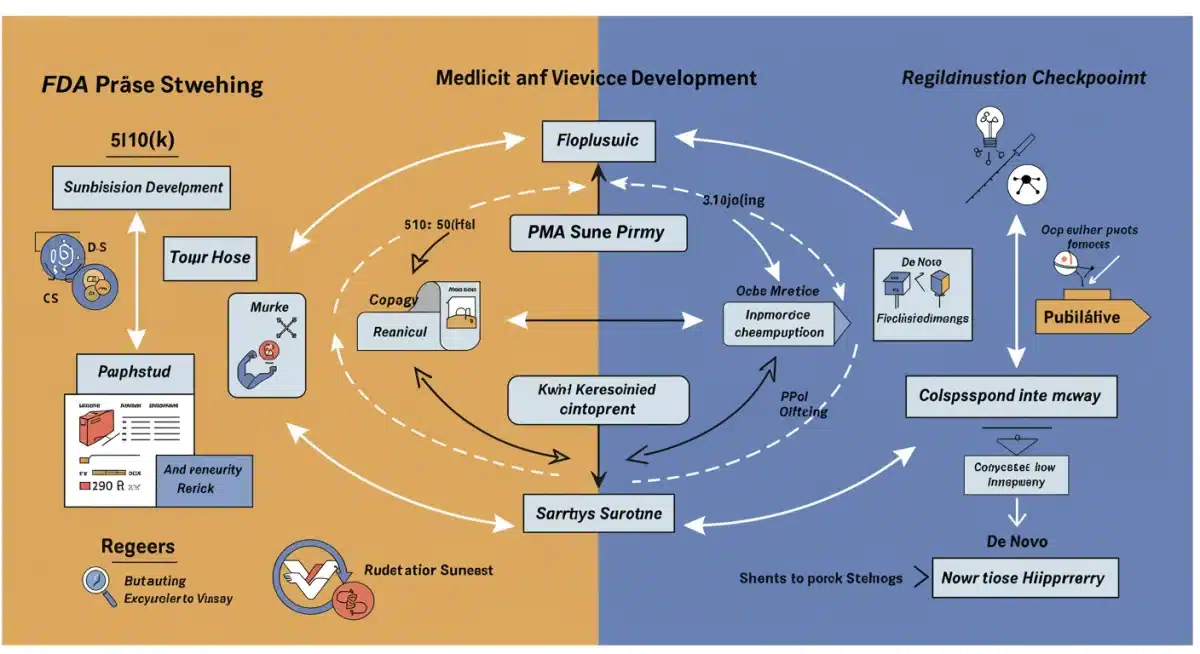
Pre-Market Submissions: Navigating the Pathways to Approval
The journey of a medical device from concept to market is heavily reliant on successful pre-market submissions. The FDA MedTech Regulations 2026 will continue to refine these pathways, with a particular focus on efficiency, clarity, and the rigorous assessment of novel technologies. US professionals must be adept at choosing the correct submission type and preparing comprehensive, high-quality documentation.
Refinements in Submission Types
- 510(k) Pre-market Notification: This remains the most common pathway for devices substantially equivalent to a predicate device. The FDA is emphasizing clearer comparisons to predicates, robust performance data, and comprehensive risk assessments, especially for devices incorporating new materials or software features.
- PMA (Pre-market Approval): For Class III devices that pose the highest risk, PMAs require extensive clinical data to demonstrate safety and effectiveness. Expect continued scrutiny on clinical trial design, statistical analysis, and the inclusion of diverse patient populations.
- De Novo Classification Request: This pathway is for novel low-to-moderate risk devices for which no predicate exists. The FDA is encouraging early engagement for De Novo submissions, offering guidance to streamline the process for truly innovative technologies.
- Breakthrough Devices Program: This program aims to expedite the development and review of certain medical devices that provide more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions. Companies should leverage this program when applicable, understanding its specific data requirements and interactive review process.
The FDA is also investing in digital tools and guidance documents to assist manufacturers in preparing high-quality submissions. Leveraging these resources can significantly reduce review times and the likelihood of requests for additional information.
Quality System Regulation (QSR) and Compliance in 2026
The Quality System Regulation (QSR), 21 CFR Part 820, is the bedrock of medical device manufacturing in the US. It outlines the requirements for establishing and maintaining a quality system to ensure that devices are safe and effective. While the core principles remain, the FDA MedTech Regulations 2026 will see continued alignment with international standards, notably ISO 13485:2016, and an increased focus on specific aspects of quality management.
Harmonization with ISO 13485:2016
The FDA has been moving towards aligning 21 CFR Part 820 with ISO 13485:2016, the international standard for medical device quality management systems. This harmonization is expected to simplify compliance for manufacturers operating in multiple global markets. While the exact timeline for full adoption as a recognized standard in the US may vary, companies should proactively transition their QMS to meet ISO 13485 requirements, if they haven’t already. This includes:
- Risk-Based Approach: Integrating risk management throughout the entire product lifecycle, from design and development to production and post-market activities.
- Software Validation: Enhanced requirements for validating software used in production, quality system processes, and as a medical device itself (SaMD).
- Supplier Control: More rigorous controls over suppliers and outsourced processes, ensuring that quality expectations are flowed down and monitored effectively.
- Post-Market Surveillance Integration: Tighter integration of post-market surveillance data into the quality system to drive continuous improvement and address emerging safety concerns.
Enhanced Focus Areas
Beyond harmonization, the FDA will likely increase its scrutiny on:
- Design Control: Ensuring robust design input, output, review, verification, and validation processes, especially for complex devices and those incorporating AI/ML.
- Process Validation: Verifying that manufacturing processes consistently produce devices meeting specifications.
- Corrective and Preventive Actions (CAPA): The effectiveness of CAPA systems in identifying, investigating, and correcting quality problems to prevent recurrence.
- Management Responsibility: The active involvement and commitment of top management in maintaining an effective quality system.
A proactive approach to QSR compliance, anticipating these enhanced focus areas, will be critical for avoiding costly non-conformances and ensuring uninterrupted market access.
Post-Market Surveillance and Real-World Evidence
The FDA’s regulatory oversight doesn’t end with market clearance. Post-market surveillance is crucial for identifying rare adverse events, understanding long-term device performance, and detecting unanticipated risks. The FDA MedTech Regulations 2026 will deepen the reliance on real-world evidence (RWE) and advanced analytical techniques to enhance post-market safety monitoring.
Leveraging Real-World Evidence (RWE)
RWE, derived from real-world data (RWD), is increasingly being used to support regulatory decisions throughout the device lifecycle. In 2026, expect the FDA to:
- Expand RWE Use Cases: Beyond post-market studies, RWE may be used to support pre-market submissions, expand indications for use, or fulfill post-approval study requirements.
- Provide Clearer Guidance: The FDA will likely issue more specific guidance on the types of RWD that are acceptable, methodologies for RWE generation, and standards for data quality and integrity.
- Promote Data Linkage: Encourage the linkage of various RWD sources (e.g., EHRs, claims data, device registries) to create more comprehensive and robust evidence.
Strengthening Adverse Event Reporting
Mandatory adverse event reporting remains a cornerstone of post-market surveillance. The FDA continues to refine its reporting systems (e.g., MAUDE database) and expects manufacturers to:
- Ensure Timely Reporting: Adhere strictly to timelines for reporting serious injuries, deaths, and malfunctions.
- Conduct Thorough Investigations: Fully investigate reported events to determine root causes and implement appropriate corrective actions.
- Utilize Data Analytics: Employ advanced analytics to identify trends, potential safety signals, and emerging risks from adverse event data.
A robust post-market surveillance plan, integrated with a strong quality system and the capacity to generate and analyze RWE, will be essential for continuous compliance and proactive risk management.
Cybersecurity in Medical Devices: A Non-Negotiable Imperative
As medical devices become more interconnected and integral to patient care, the threat of cyberattacks grows exponentially. The FDA MedTech Regulations 2026 will solidify cybersecurity as a critical component of device safety and effectiveness, requiring a comprehensive approach from design to end-of-life.
Pre-Market Cybersecurity Requirements
Manufacturers must demonstrate robust cybersecurity measures in their pre-market submissions. This includes:
- Cybersecurity Bill of Materials (CBOM): Providing a list of all commercial, open-source, and off-the-shelf software and hardware components, including their versions and known vulnerabilities.
- Threat Modeling and Risk Assessment: Conducting thorough threat modeling and cybersecurity risk assessments to identify potential vulnerabilities and their impact.
- Secure Design Principles: Incorporating security by design, including authentication, authorization, data encryption, and secure update mechanisms.
- Cybersecurity Testing: Performing rigorous testing, including penetration testing and vulnerability scanning, to validate device security.
- Cybersecurity Management Plan: Submitting a plan for ongoing cybersecurity management throughout the device’s lifecycle, including patching, updates, and incident response.
Post-Market Cybersecurity Management
The FDA also expects manufacturers to maintain proactive post-market cybersecurity vigilance:
- Vulnerability Monitoring: Continuously monitoring for new vulnerabilities in their devices and their components.
- Patching and Updates: Developing and deploying patches and updates in a timely manner to address identified vulnerabilities.
- Incident Response Plan: Having a well-defined and tested plan for responding to cybersecurity incidents, including communication with the FDA and affected users.
- Information Sharing: Participating in information sharing organizations (e.g., H-ISAC) to stay informed about emerging threats and best practices.
Failure to adequately address cybersecurity risks can lead to significant regulatory actions, reputational damage, and, most importantly, patient harm. Cybersecurity is no longer an IT concern; it’s a core regulatory and patient safety issue.
Digital Health Technologies (DHTs) and Artificial Intelligence (AI)/Machine Learning (ML)
The rise of digital health technologies (DHTs) and the integration of AI/ML into medical devices represent a transformative shift in healthcare. The FDA MedTech Regulations 2026 are particularly focused on providing clear regulatory pathways and oversight for these complex and rapidly evolving innovations.
Software as a Medical Device (SaMD)
SaMD, defined as software intended to be used for one or more medical purposes without being part of a hardware medical device, presents unique regulatory challenges. The FDA is refining its approach to SaMD through:
- Pre-Cert Program (Potential): While the Pre-Cert program is still under development and evaluation, its principles of assessing organizational excellence rather than product-by-product review may influence future SaMD regulation.
- Risk-Based Categorization: Clearer guidance on categorizing SaMD based on its intended use and the criticality of the information it provides to healthcare decisions.
- Clinical Validation: Emphasizing the need for robust clinical validation to demonstrate the accuracy, reliability, and clinical benefit of SaMD.
AI/ML-Driven Medical Devices
AI/ML algorithms can adapt and learn over time, posing challenges to traditional fixed-functionality regulatory models. The FDA’s approach to AI/ML-driven devices includes:
- Total Product Lifecycle (TPLC) Approach: A framework that considers the entire lifecycle of AI/ML devices, from pre-market development to post-market performance monitoring and algorithm changes.
- Good Machine Learning Practice (GMLP): Principles for developing high-quality, transparent, and robust AI/ML algorithms, focusing on data management, model development, and performance monitoring.
- Predetermined Change Control Plans (PCCPs): For adaptive AI/ML devices, manufacturers may be able to submit PCCPs that outline the types of modifications that can be made to the algorithm without requiring a new 510(k) or PMA, provided certain guardrails are met.
- Transparency and Explainability: Increasing expectations for manufacturers to provide clear explanations of how AI/ML algorithms work, their limitations, and how they contribute to clinical decision-making.
Engaging with the FDA early for novel DHTs and AI/ML devices is highly recommended to leverage existing guidance and potentially influence future policy development.
Supply Chain Resiliency and Transparency
The COVID-19 pandemic exposed vulnerabilities in global medical device supply chains. In response, the FDA MedTech Regulations 2026 will likely reinforce requirements for supply chain resiliency, transparency, and quality management to prevent future disruptions.
Enhanced Supplier Management
Manufacturers will face increased scrutiny on their supplier management programs, including:
- Supplier Qualification: More rigorous processes for qualifying and auditing suppliers, especially those providing critical components or services.
- Quality Agreements: Comprehensive quality agreements that clearly define responsibilities, quality standards, and communication protocols with suppliers.
- Risk-Based Monitoring: Implementing risk-based monitoring strategies for suppliers, focusing on those with the highest impact on product quality and availability.
Supply Chain Visibility and Traceability
The FDA is pushing for greater visibility and traceability throughout the supply chain:
- Component Traceability: The ability to trace critical components from their origin to the finished device.
- Geographic Diversity: Encouraging manufacturers to diversify their supply chains geographically to reduce reliance on single regions.
- Contingency Planning: Requiring robust contingency plans for supply chain disruptions, including alternative suppliers and manufacturing sites.
Proactive mapping of supply chains, identifying critical nodes, and developing mitigation strategies will be crucial for maintaining product availability and regulatory compliance.
Preparing for Success: Actionable Strategies for US Professionals
Navigating the complex and evolving FDA MedTech Regulations 2026 requires a strategic and proactive approach. Here are actionable steps US professionals can take to ensure their organizations are well-prepared:
- Stay Informed and Educated: Regularly monitor FDA guidance documents, workshops, and public meetings. Subscribe to FDA newsletters and engage with industry associations (e.g., AdvaMed, RAPS) for the latest updates. Continuous professional development in regulatory affairs is non-negotiable.
- Conduct a Regulatory Gap Analysis: Proactively assess your current quality system, design controls, and post-market surveillance processes against the anticipated 2026 requirements. Identify any gaps and develop a remediation plan.
- Invest in Cybersecurity Infrastructure and Expertise: Elevate cybersecurity from an IT function to a core business and regulatory imperative. Invest in skilled personnel, advanced security tools, and robust incident response planning.
- Embrace a Risk-Based Approach: Integrate risk management principles into every stage of the product lifecycle, from initial concept to post-market surveillance. This aligns with the FDA’s increasing emphasis on proactive risk identification and mitigation.
- Leverage Digital Tools for Compliance: Explore and implement digital solutions for quality management, document control, adverse event reporting, and data analytics. These tools can enhance efficiency, accuracy, and traceability, which are vital for compliance.
- Foster Cross-Functional Collaboration: Regulatory compliance is not solely the responsibility of the regulatory affairs department. Encourage seamless collaboration between R&D, engineering, quality, manufacturing, clinical, and marketing teams to embed regulatory requirements throughout the organization.
- Engage Early with the FDA: For novel devices, particularly those involving AI/ML or complex digital health components, consider early engagement with the FDA through Q-Submission (Pre-Submission) meetings. This can provide valuable feedback and clarify regulatory expectations.
- Strengthen Supplier Quality Management: Re-evaluate and strengthen your supplier qualification, auditing, and monitoring processes. Ensure clear quality agreements are in place and that your supply chain is resilient to disruptions.
- Prioritize Training and Awareness: Ensure all relevant personnel receive adequate training on the latest FDA regulations and internal quality procedures. A well-trained workforce is your first line of defense against compliance issues.
- Develop a Culture of Quality and Compliance: Ultimately, regulatory success stems from an organizational culture that prioritizes quality, patient safety, and ethical conduct. Leadership commitment and consistent reinforcement are key.
Conclusion
The FDA MedTech Regulations 2026 represent a continued evolution of the regulatory framework designed to ensure the safety and effectiveness of medical devices in a rapidly advancing technological landscape. For US professionals in the MedTech industry, understanding and proactively adapting to these changes is not just about avoiding penalties; it’s about fostering innovation, maintaining market access, and ultimately, safeguarding public health.
By embracing a forward-thinking approach, investing in robust quality systems, prioritizing cybersecurity, and leveraging digital health advancements responsibly, companies can navigate the complexities of the 2026 regulatory environment with confidence. The future of MedTech is bright, but its path is inextricably linked to diligent adherence to regulatory requirements and a steadfast commitment to excellence. Stay informed, stay compliant, and continue to innovate for a healthier future.





